


Contact Info
Adam P. Hitchcock
Canada Research Chair
in Materials Research
CLS-CCRS
B.I.M.R
McMaster University
Hamilton, ON
Canada L8S 4M1
V: +1 905 525-9140
x24729
F: +1 905 521-2773
E: aph@mcmaster.ca
U: unicorn.mcmaster.ca
__________
Research
Group
Opportunities
Publications
Links
_____________

|
|
PHYSICS in CANADA 55 (1999) 191-198 (September/October, 1999)
Applications of synchrotron light to physics and chemistry
Adam P. Hitchcock
Dept. of Chemistry, & Brockhouse Institute for Materials Research
McMaster University, Hamilton, ON Canada L8S 4M1
John J. Neville
Dept. of Chemistry, University of New Brunswick,
Box #45222, Fredericton, NB Canada E3B 6E2
Abstract
Synchrotron light is an accelerator based light source that covers the
complete electromagnetic spectrum from infrared to hard X-ray. It is intense,
bright, fully tunable, highly polarized, and has a time structure useful
for many types of dynamics experiments. Aside from the visible, near-IR
and near-UV where lasers are generally superior, the properties of synchrotron
light far surpass those of available lab sources. The ~50 facilities world
wide are used by up to 20,000 scientists annually, from many disciplines,
but researchers from physics and chemistry are the most prevalent. This
article describes some Canadian research in this area using mainly examples
from our research. It also describes the Canadian Light source, a proposed
2.9 GeV facility that will enable Canadian scientists to contribute more
effectively in this arena. The future of synchrotron research in Canada
is bright indeed!
1. What is synchrotron light ?
2. Research in chemistry and physics using synchrotron light
2.A Spectroscopy & Dynamics
Synchrotron light is virtually unique in its ability to tune photon energy with continuous coverage over wide ranges and with excellent intensity. This leads to many different types of applications in spectroscopy - mapping the quantized energy levels of a gas, liquid or solid target and using these results to learn about the electronic and geometric structure of the sample. Much of the spectroscopy with synchrotron light deals with inner-shell or core levels for which energies above 50 eV are required. Mike Bancroft (Western) has spent much of his career working on systematic investigations of the X-ray photoelectron and X-ray photoabsorption spectroscopy of inorganic compounds with the Canadian Synchrotron Radiation Facility, several beamlines at the Synchrotron Radiation Centre (Madison, WI ) [4,5] High resolution spectroscopy of various types is an important field of chemical physics, one in which Canada is very active. Synchrotron spectroscopy studies are continuing this tradition. For example. John Hepburn (Waterloo) is carrying out high resolution spectroscopy [6] and photoionization dynamics measurements in the vacuum ultraviolet range at the Advanced Light Source (Berkeley, CA).
Photoionization dynamics investigates the time evolution of specific
states of atoms, molecules or solids. Photoionization, and related processes
such as X-ray or Auger decay of core holes, provide a powerful tool to
study dynamics, either directly through combined laser-synchrotron pump-probe
approaches - such as are being carried out by Larzillière (Laval)
at Super-ACO, Paris - or indirectly, using the characteristic time-scale
of the core hole decay as a 'femto-second clock'. An example of the latter
is the phenomena of ultra-fast decay whereby a bond in a molecule is broken
in the core excited state and dissociation occurs before the Auger process
refills the core hole. Aksela, Bancroft and coworkers [7] were
one of the first to document the ultra-fast decay process in the dissociative
(Cl2p-1,s *) state of HCl. Recently
X-ray emission using fully resolved incident and outgoing photons has
been shown to be an extremely powerful way to study photoionization dynamics.
At the deepest level one needs to make a unified description of the excited
state creation, its time evolution, and its subsequent decay. This new
understanding has implications for photochemical control - for example,
it has been shown that tuning the incident photon energy away from a core
excitation resonance can change the nature of the core-valence coupling
and thus the pathway of the decay [8]. 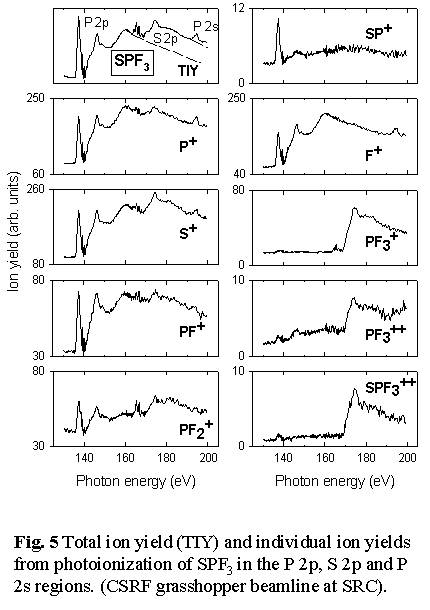
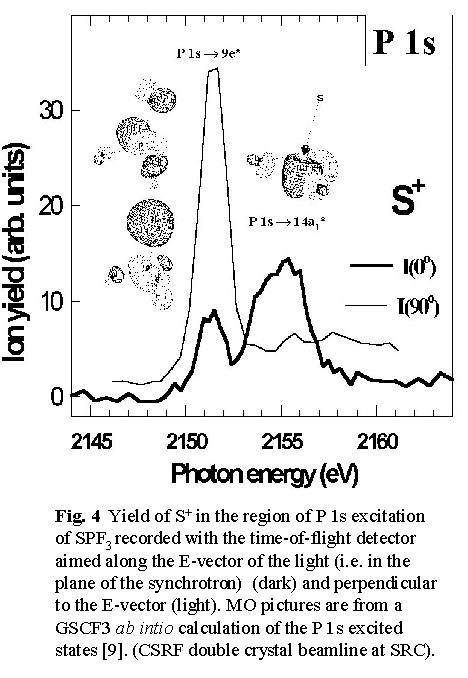
We are using time-of-flight mass spectrometry and multi-stop coincidence techniques at the CSRF beamlines at SRC to investigate the core-excitation spectroscopy and dynamics of core state decay of small molecules. Fig, 4 shows a spectroscopy example from a recent study of SPF3 [9] in which the symmetry of core excited states is determined directly from the angular distribution of ionic fragmentation processes which lead to pairs (or triplets) of energetic ions. Basically, if bond breaking in an energetic ionic fragmentation occurs before the molecular orientation has had a chance to randomize by rotation, then the direction of preferential ion emission is closely related to the symmetry of the excitation. In this case, when the time-of-flight detector is aligned perpendicular to the electric vector of the light the P 1s ® 9e* transition dominates the S+ partial ion yield spectrum, whereas when the time-of-flight detector is rotated 90o to place the time-of-flight detector parallel to the electric vector, the P 1s ® 14a1* transition becomes strongest. By comparison, the total ion yield spectra and partial yield spectra of thermal ions from P 1s excited SPF3 show negligible angular anisotropy. Fig 4 also includes sketches of the molecular orbitals involved in these core excitations. The diagrams are derived from ab initio calculations of core excited states. Parallel experimental and computational studies are an integral part of synchrotron science, as in almost all other areas of chemistry and physics.
Auger decay of inner-shell excited and ionized states is an efficient
source of multiply charged ions. The charge separation and fragmentation
of these species, studied by photoelectron-photoion-photoion coincidence
(also called charge separation mass spectrometry [10]) gives insights
into bonding and electronic structure. In molecules, the dependence of
the fragmentation process on the X-ray energy can reveal cases of site
and/or state selective fragmentation.
The identification of potential candidates for selective X-ray photochemistry
is an ongoing goal of our research program. In addition to being an aspect
of photoionization dynamics, state-selective photochemistry in either
gas or condensed phases can be used to control chemical reactions in a
way that may be useful for making new materials or devices. Comparison
of the different core edge signals in SPF3 reveals that this
molecule is one with a high degree of site-selective fragmentation [11]
- see Figs. 5 and Fig. 6. 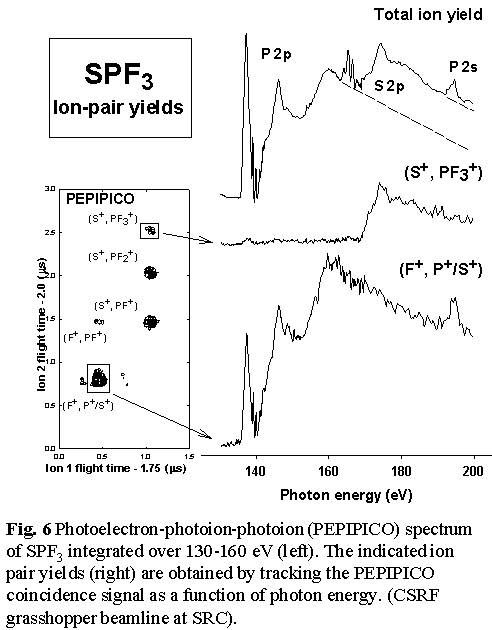 Thus
loss of all 3 fluorine atoms to produce SP+ occurs predominantly
with P 2p excitation, and not at all with S 2p excitation or ionization.
In contrast, the S=P bond is much more readily broken with S 2p excitation,
as evidenced by the production of PF3+, PF3++
and the (S+,PF3+) ion pair signals only
following S 2p excitation. Even complete dissociation of the molecule
into ionized atoms appears to have site selectivity since the (F+,S+/P+)
atomic ion pair signal is exclusively generated by P 2p/2s excitation
and ionization [11].
Thus
loss of all 3 fluorine atoms to produce SP+ occurs predominantly
with P 2p excitation, and not at all with S 2p excitation or ionization.
In contrast, the S=P bond is much more readily broken with S 2p excitation,
as evidenced by the production of PF3+, PF3++
and the (S+,PF3+) ion pair signals only
following S 2p excitation. Even complete dissociation of the molecule
into ionized atoms appears to have site selectivity since the (F+,S+/P+)
atomic ion pair signal is exclusively generated by P 2p/2s excitation
and ionization [11].
Similar investigations of photoionization dynamics of core states are being carried out by a number of groups with Canadian participation, including studies by Chris Brion (UBC) and Tong Leung (Waterloo). Recently Leung, in collaboration with Lindle and coworkers [12] have discovered that neutral hydrogen is generated in high yield in Cl 1s excited HCl and S 1s excited H2S. While the origin is not fully understood, one possibility is ultrafast decay - bond breaking prior to core hole decay. If correct, this would require very fast neutral separation since the core hole lifetime is extremely short for such a high energy state.
Analysis of the peak shapes from multi-ion coincidence signals can provide
detailed information about energetics, angular distributions, and thus
fragmentation mechanisms. Fig. 7 shows the line shape of the three
ion pair signals - (F+,PF2+), (F+,PF+)
and (F+,P+) - produced from P 2p ionization of PF3.
The peak at the upper left of the lower two contour plots arises from
events in which the lighter ion (F+) flies directly towards
the time-of-flight detector and the heavier ion (PFx+,
x= 0-2) flies directly away, while the peak to the lower right 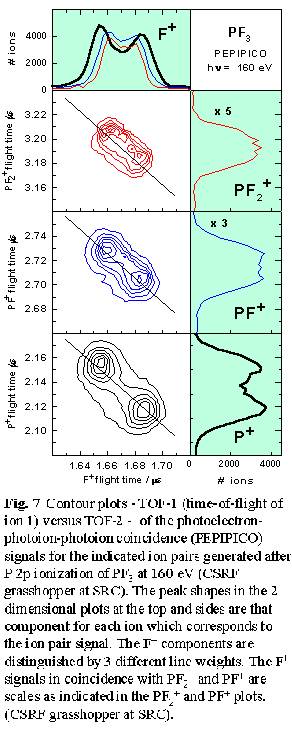 corresponds
to the opposite process. The width of the (F+,PF2+)
signal or the peak separation is determined by the kinetic energy released.
The average kinetic energy, along with the shape and alignment of the
coincidence signal on the TOF-1 versus TOF-2 plot, gives useful insights
into fragmentation mechanisms. In this case the (F+,PF2+)
signal is aligned exactly along the -1 diagonal (as is required for a
two body dissociation [12]), with a size indicating an average kinetic
energy release of 9.0 eV, consistent with the 9.2 eV energy of Coulomb
repulsion of two ions separated by the P-F bond length. The undetected
neutral or ion partners generated in events leading to (F+,PF+)
and (F+,P+) pair signals can take away energy and
momentum and thus it is possible these signals are associated with fragmentation
processes with mechanisms very different from a simple two-body concerted
dissociation. Surprisingly, the orientation (slopes close to -1) and line
shape of the (F+,PF+) and (F+,P+)
ion pair signals are consistent with these ion pairs being produced in
concerted dissociative processes, with most of the kinetic energy taken
by these two ions. However, for (F+,P+) the average
kinetic energy release (14.4 eV) is significantly higher than the 9.2
eV Coulomb repulsion value. Even if the other two F atoms formed F2
this would provide only 1.6 eV, much less than the excess 5.2 eV.
From this it appears a significant portion of the (F+,P+) pair signal
may arise from triple ionization processes in which a second F+
is generated but not detected since its flight time would be nearly identical
to that of the detected F+ ion.
corresponds
to the opposite process. The width of the (F+,PF2+)
signal or the peak separation is determined by the kinetic energy released.
The average kinetic energy, along with the shape and alignment of the
coincidence signal on the TOF-1 versus TOF-2 plot, gives useful insights
into fragmentation mechanisms. In this case the (F+,PF2+)
signal is aligned exactly along the -1 diagonal (as is required for a
two body dissociation [12]), with a size indicating an average kinetic
energy release of 9.0 eV, consistent with the 9.2 eV energy of Coulomb
repulsion of two ions separated by the P-F bond length. The undetected
neutral or ion partners generated in events leading to (F+,PF+)
and (F+,P+) pair signals can take away energy and
momentum and thus it is possible these signals are associated with fragmentation
processes with mechanisms very different from a simple two-body concerted
dissociation. Surprisingly, the orientation (slopes close to -1) and line
shape of the (F+,PF+) and (F+,P+)
ion pair signals are consistent with these ion pairs being produced in
concerted dissociative processes, with most of the kinetic energy taken
by these two ions. However, for (F+,P+) the average
kinetic energy release (14.4 eV) is significantly higher than the 9.2
eV Coulomb repulsion value. Even if the other two F atoms formed F2
this would provide only 1.6 eV, much less than the excess 5.2 eV.
From this it appears a significant portion of the (F+,P+) pair signal
may arise from triple ionization processes in which a second F+
is generated but not detected since its flight time would be nearly identical
to that of the detected F+ ion.
2.B Materials Science
Many of the applications of synchrotron light are in the area of high performance analysis of new materials. There is an old saying, 'you can't control what you can't see'. By pushing the frontiers of what can be analyzed - lowering spatial, temporal, precision, and detection limits - synchrotron light is contributing in diverse and significant ways to new material and new device development. By the year 2003 it is widely expected that semiconductor devices requiring the finest features - particularly microprocessors and high capacity memory chips - will use X-ray lithography based on synchrotron light in part of their manufacture. Total reflection X-ray fluorescence analysis - a photon in - photon out technique - is being developed as a key technique to monitor contamination of semiconductor surfaces, an issue which becomes increasingly critical as device dimensions shrink.
In the area of analytical sciences, synchrotron light is greatly enhancing the capabilities of well-established techniques such as X-ray photoelectron spectroscopy, and also leading to the development of new techniques, such as resonant inelastic X-ray scattering [13], zone-plate based X-ray spectromicroscopy [14], and extended X-ray absorption fine structure (XAFS) [2,15]. Some of our recent work in zone plate microscopy is described below to illustrate only one aspect of the exciting new materials science enabled by synchrotron light.
2.B.1 Microscopy 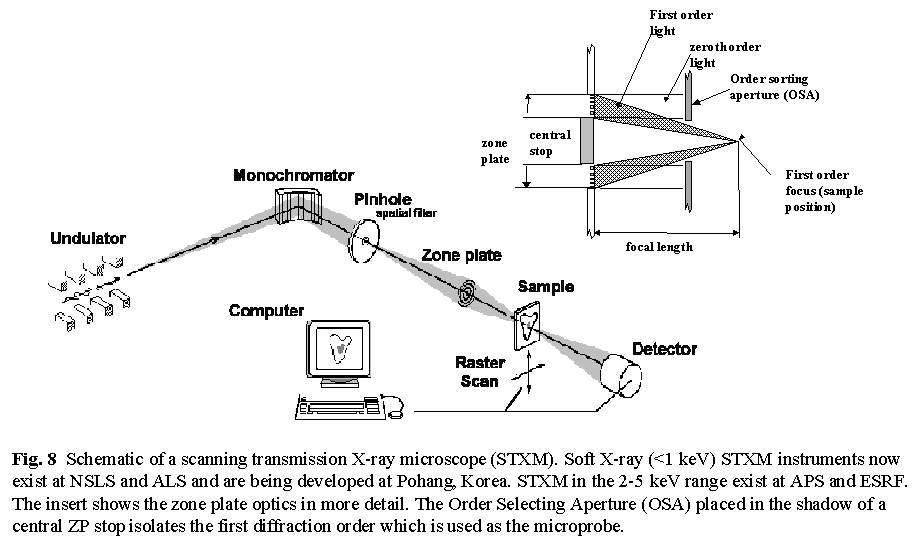
Fig. 8 shows a schematic of a scanning transmission X-ray microscope (STXM) [14]. It uses a small X-ray spot created by focussing monochromatized x-rays with a Bragg-Fresnel zone plate, which is a circular transmission diffraction grating with a variable line spacing. The zone plate uses only the spatially coherent component of the X-rays with which it is illuminated so the high brightness and partial coherence of third generation undulator light is critical to their performance. In addition, the fabrication of soft X-ray zone plates pushes the limits of modern electron beam mask generation and lithographic techniques so the spatial resolution is closely related to how fine and precise the zone plate structure can be made. Images are produced pixel by pixel by raster scanning the sample through the beam while recording a suitable signal such as transmitted flux (STXM), fluorescent X-rays, luminescence, or photo-ejected electrons (SPEM) In STXM the sample is located in an atmospheric pressure helium enclosure, a vacuum, or in a liquid contained within X-ray transparent windows. Imaging with the photon energy tuned to chemically-specific spectral features provides chemical maps. Alternatively, the photon energy can be scanned to acquire a Near Edge X-ray Absorption Fine Structure (NEXAFS) spectrum from a small area. Recently procedures to acquire sequences of full images at many photon energies have been implemented (see core-shell particles, toner particles, protein-on-polymers, or jacobsen web site for examples). The acquisition of an image sequence is an extremely powerful way of studying samples which has already provided many interesting surprises, revealing aspects of sample chemistry that would not have been discovered by other means.
So what is new ? - what does X-ray spectromicroscopy offer, that
can't be provided by other techniques such as high performance optical
or electron microscopy? Even the best optical techniques have an order
of magnitude lower spatial resolution (~0.5 m
m versus 50 nm) and, unless some intrusive chemistry is employed, such
as species-specific fluorescent labeling, optical techniques have much
less chemical sensitivity. Electron microscopy has better spatial resolution
(as good as 0.1 nm), but there are severe limitations associated with
radiation damage when studying sensitive polymer or biological samples.
We have demonstrated [16] that NEXAFS microscopy has an advantage of about
three orders of magnitude over the corresponding electron microscopy technique
in being able to spectroscopically analyze and image small sample areas
of radiation sensitive materials such as polymers. In addition, x-ray
linear dichroism microscopy can determine orientation of chemical moieties
at high spatial resolution [17]. These inherent advantages of x-ray microscopy
have opened up new avenues for the characterization of polymers.
In collaboration with John Dutcher (Physics, Guelph) we
are using X-ray microscopy to investigate mechanisms of polymer "self-organization".
Patterning of thin polymer films is attracting increasing interest, both
from a fundamental point of view, and on account of potential technological
applications. Normally polymer films are deposited on a flat substrate
such as a silicon wafer. However the thermal stability of deposited films
can be a limitation on account of hole formation caused by dispersion
forces which amplify structural instabilities at the film surface. Mechanical
confinement of polymer films offers the possibility to control this instability
which raises possibilities for unique self-assemblies of polymers on surfaces
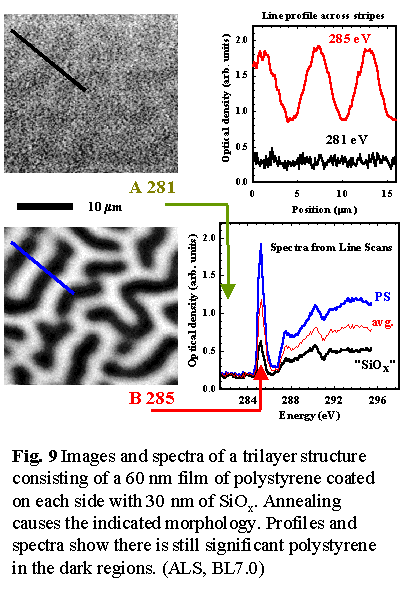
patterned on submicron length scales. We have used STXM to study very
thin (~50 nm prior to annealing) polystyrene films confined between thin
continuous layers of silicon suboxide (see Fig. 9). The SiOx
layers prevent hole formation. However aggressive annealing above the
glass transition temperature (Tg) generates a novel structure
driven not by phase segregation, but by the attractive dispersion force
between the SiOx-air surface [18]. These structures offer an
attractive test-bed for understanding and developing control mechanisms
for one of the fundamental forces governing self-assembly of polymer structures.
In order to fully model the mechanisms for this spontaneously generated
lateral morphology, it is important to know the thickness of the trapped
polymer film throughout the structure. STXM readily provides this information.
Prior to these measurements it had been assumed that there was almost
complete exclusion of the polystyrene from the dark, non-continuous regions.
However, STXM indicated ~30% residual polystyrene in those areas (see
Fig. 9). In the near future we plan to investigate trapped thin film polymers
of materials with different strengths of interaction with SiOx.
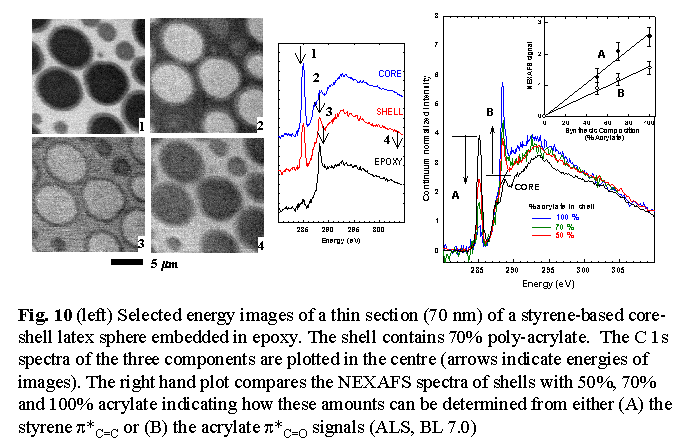
A second example is from recent STXM studies of novel latex particles
with internal structure produced by Harald Stöver's group
(BIMR, Chemistry, McMaster). These latex spheres are attractive for a
number of applications, including coatings, adhesives, chemical delivery
etc. In order to optimize these structures for particular applications
(e.g. pheromone delivery for environmentally benign, controlled release
pest control) it is important to be able to determine the origin of their
internal structure. In particular, while electron microscopy of cross-sections
of latex spheres readily reveals the presence or absence of internal structure
of micron sized latex particles, the limited chemical sensitivity of electron
microscopy means that one cannot be sure if the observed structure arises
from a genuine chemical difference in the various regions (e.g. core versus
shell) or whether it is a consequence of a density and/or thickness variation
(perhaps driven by some underlying chemical difference).

Comparison of STXM images at several photon energies provide a
means to quantify the relative contributions of chemical versus density/thickness
contrast. Fig 10 shows examples of a recent study of acrylate-polystyrene
latex particles which have been synthesized in a two-step process to intentionally
segregate all of the poly-acrylate component to the shell region. The
exquisite sensitivity of NEXAFS microscopy to this structure is clear.
Quantitative chemical compositions of the shell can be derived from the
intensities of specific features of the NEXAFS spectra of the shell region
(see Fig. 10). More challenging has been a study of a 'tree-ring' morphology
in latex particles formed from a blend of styrene and chlorostyrene (Fig
11). The 'tree-ring' morphology is observed only at specific photon
energies. This clearly points to a chemical compositional origin rather
than a thickness or density origin of this morphology. In turn, this suggests
the tree-ring morphology is generated by heterogeneous chemical oscillations
in the one-step synthesis.
3. Canadian Light Source
In 1990 many scientists in Canada grouped together to form the Canadian Institute for Synchrotron Radiation (CISR), an umbrella organization dedicated to education, co-ordination, and facilitation of synchrotron light research. There are currently about 350 faculty level members, more than half of whom are directing research programs at off-shore facilities. In part through CISR initiatives, there is now formal Canadian participation in one or more beam line projects at ALS (Berkeley), APS (Argonne), and SRC (Madison).
Until the start of the CLS project, Canada was virtually the only developed nation without a synchrotron light facility. In 1994 a committee of NSERC identified the need for a light source in Canada. This led to an open competition, in which groups from Eastern Canada and Western Canada competed with site specific project proposals. In 1996 a peer review committee, administered by NSERC and chaired by Dr. Alex McAuley (Victoria) re-affirmed the need for a light source and recommended that Saskatoon be the site. NSERC council accepted the recommendations of the McAuley committee in Nov 1996. In 1999, with the award of a Canada Foundation for Innovation grant for $54 M, the Canadian Light Source project was launched. The toal project budget is ovr $140 M in new funds (and over $170 M if the value of the infrastructure inherited from the SAL faiclity is included). The first scientific experiments with CLS generated synchrotron light will occur in 2004.
CLS will be a full service national synchrotron light facility. The 2.9 GeV, low emittance CLS design is fully competitive third generation technology, with high current (500 mA in 2008). It will have a brightness comparable to existing third generation facilities (see Table 1). When equipped with a small gap undulator, the CLS will even be competitive with wigglers at the Advanced Photon Source. Relative to other third generation facilities the CLS design is characterized as having the highest beam energy and the highest brightness per unit accelerator length. Thus there are novel aspects of the machine design and the research program that will make CLS truly unique. The CLS team is confident that CLS can be built on time and on budget.
Table 1. Comparison of the ring energy and emittance parameters (critical to brightness of the light) for ALS, CLS and APS

In developing plans for the CLS the Canadian synchrotron light community identified user communities and a documentable need for 12 light points (3 undulator, 2 wiggler, and 7 bending magnet lines) with a total of 15 end stations. The CLS has an ultimate capacity for up to 48 bending magnet lines and 10 insertion device lines, allowing more than 50 different experiments to be carried out simultaneously. Matching funds for approximately half of the cost of each of the initial lines have been committed by the federal and Saskatchewan provincial governments. User groups for each research area are in the process of identifying sources, such as other provincial capital development funds or industrial partners, of the remaining funds needed to complete the first phase CLS beam lines.
4. How can you learn more about synchrotron light research ?
If you are interested in a specific topic discussed in this article contact one of the authors or the principal investigator identified in the text. Web sites which provide further information of specific organizations or synchrotron light facilities are listed in table 2.
Table 2. Institutions and contact information
| Institute/facility | location | Web site |
| CISR - Canadian Institute for Synchrotron Radiation | UWO, London, ON Saskatoon, SK | www.cisr.ca |
| CLS - Canadian Light Source | Saskatoon, SK | cls-ccrs.usask.ca |
| ALS - Advanced Light Source | Berkeley, CA | www-als.lbl.gov |
| APS - Advanced Photon Source | Argonne, IL | www.aps.anl.gov |
| NSLS - National Synchrotron Light Source | Brookhaven, NY | www.nsls.bnl.gov |
| SRC - Synchrotron Radiation Centre | Madison, WI | www.src.wisc.edu |
Acknowledgements
This article has touched on the work of only a few of the more than 100
groups in Canada who are using synchrotron light. We apologize in advance
for our restricted selection of examples. We would especially like to
thank our colleagues, Dr. Tolek Tyliszczak and Dr. Stephen Urquhart, and
numerous collaborators, who have played a major role in the synchrotron
chemical physics research described herein.
References
- H. Winick (ed), Synchrotron Radiation Sources: A primer, (World Scientific, 1994).
- J. Stöhr, NEXAFS Spectroscopy (Springer, 1992), D.C. Koningsberger, R.Prins (eds), X-ray Absorption (Wiley, 1988).
- B.H. Kear, Scientific American 255 (1986) 159; M. Sutton and G.B. Stephenson, Physics in Canada 44 (1987) 131 ; Physics Today (May, 1981, June, 1983, March, 1985)
- G.M. Bancroft, K.H. Tan and J.D. Bozek, Physics in Canada 43 (1987) 113; B.X. Yang, F. Middleton, B. Olssen, G.M. Bancroft, J.M. Chen, T.K. Sham, K.H. Tan and D. Wallace, Rev. Sci. Inst. 63, 1355 (1992).
- Z.F. Liu, G.M. Bancroft, K.H. Tan and M. Schachter, J. Electron Spectrosc. 67 (1994) 299.
- R.C. Shiell, M. Evans, S. Stinson, C.-W. Hsu, C.Y. Ng and J.W. Hepburn, Phys. Rev. Lett. 80 (1998) 472.
- H. Aksela, S. Aksela, M. Ala-Korpela, O.-P. Sairanen, M. Hotokka, G.M. Bancroft and K.H. Tan, Phys. Rev. A 41 (1990) 6000.
- A. Cesar, F. Gel'mukhanov, Y. Luo, H. Agren, P. Skytt, P. Glans, J. Guo, K. Gunnelin and J. Nordgren, J. Chem. Phys. 106 (1997) 3439.
- J.J. Neville, T. Tyliszczak, A.P. Hitchcock, A. Jürgensen and Ron G. Cavell, Chem. Phys. Lett. (1998) in press.
- J.H.D. Eland, Acc. Chem. Res. 22 (1989) 381
- A.P. Hitchcock, J.J. Neville, A. Jürgensen and R.G. Cavell, J. Electron Spectrosc. 88-91 (1998) 71
- D.L. Hanson et al. Phys. Rev. A 58 (1998) 3757.
- F. Gel'mukhanov and H. Agren, Phys. Rev. B 57 (1998) 2780.
- H.Ade (ed). J. Electron Spectroscopy 84 (1997), special issue on soft X-ray spectromicroscopy.
- Z.H. Lu, T. Tyliszczak and A.P. Hitchcock, Phys. Rev. B 58 (1998) 13820.
- E.G. Rightor, A.P. Hitchcock, H. Ade, R.D. Leapman, S.G. Urquhart, A.P. Smith, G. Mitchell, D. Fischer, H.J. Shin and T. Warwick, J. Phys. Chem. B 101 (1997) 1950
- H. Ade and B. Hsaio, Science 262 (1993) 1427; A.P Smith and H. Ade, Appl. Phys. Lett. 69 (1996) 3833.
- K. Dalnoki, B.G. Nickel and J.R. Dutcher, Phys. Rev. Lett. 82 (1999) 1486.
|
|
© 2002 A.P. Hitchcock / McMaster University
- All Rights Reserved
web site by Christopher Amis. Last updated on 04/23/2002 (aph)